10x Genomics
Visium Spatial Gene Expression
Space Ranger1.3, printed on 08/13/2025
Image Analysis Metrics
| Metric | Description |
|---|---|
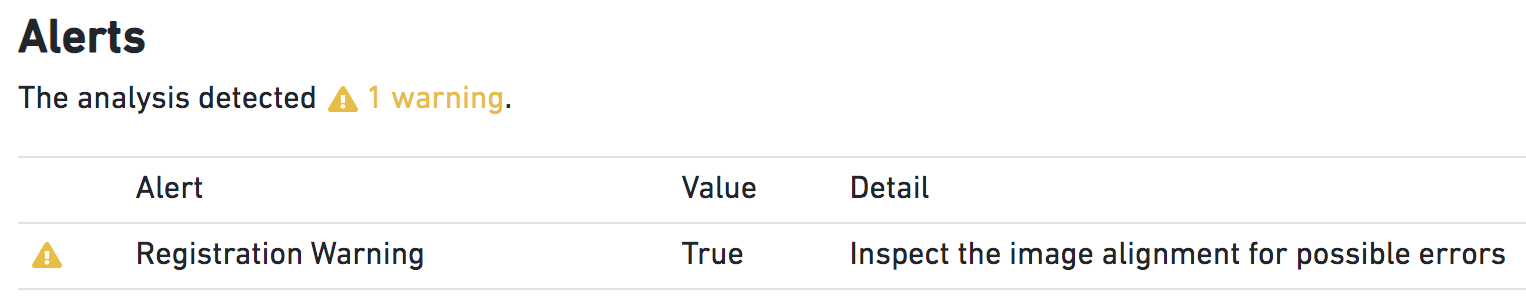
Suspect Alignment | Metric is True when the fiducial alignment algorithm fails. Alignment failure often manifests as a subtle rotation or translation that misplaces spots by a fraction of the spot diameter. These failures are difficult to automatically catch, as detection of the alignment failure is similar to the process of alignment itself. However, if the alignment process reports a scaling, rotation, or translation of the input image that seems truly implausible, the pipeline will issue a warning that the user should closely inspect the results of the fiducial alignment (see image below). The absence of such a warning is not a guarantee of success, so the user should always inspect the quality-control images from the pipeline for subtle errors. |
Gene Expression Metrics
Space Ranger pipelines output key metrics in text format. Below are the definitions of the reported metrics.
"spaceranger count" Metrics Definitions
The spaceranger count pipeline outputs metrics_summary.csv which contains a number of key metrics about the barcoding and sequencing process.
| Metric | Description |
|---|---|
Number of Spots Under Tissue | The number of barcodes associated with a spot under tissue. |
Mean Reads per Spot | The number of reads, both under and outside of tissue, divided by the number of barcodes associated with a spot under tissue. |
Median Genes per Spot | The median number of genes detected per spot under tissue-associated barcode. Detection is defined as the presence of at least 1 UMI count. |
Number of Reads | Total number of read pairs that were assigned to this library in demultiplexing. |
Valid Barcodes | Fraction of reads with barcodes that match the whitelist after barcode correction. |
Reads Mapped Confidently to Transcriptome | Fraction of reads that mapped to a unique gene in the transcriptome. The read must be consistent with annotated splice junctions. These reads are considered for UMI counting. |
Reads Mapped Confidently to Exonic Regions | Fraction of reads that mapped uniquely to an exonic region of the genome. |
Reads Mapped Confidently to Intronic Regions | Fraction of reads that mapped uniquely to an intronic region of the genome. |
Reads Mapped Confidently to Intergenic Regions | Fraction of reads that mapped uniquely to an intergenic region of the genome. |
Sequencing Saturation | The fraction of reads originating from an already-observed UMI. This is a function of library complexity and sequencing depth. More specifically, this is the fraction of confidently mapped, valid spot-barcode, valid UMI reads that had a non-unique (spot-barcode, UMI, gene). |
Q30 Bases in Barcode | Fraction of spot barcode bases with Q-score >= 30, excluding very low quality/no-call (Q <= 2) bases from the denominator. |
Q30 Bases in RNA Read | Fraction of RNA read bases with Q-score >= 30, excluding very low quality/no-call (Q <= 2) bases from the denominator. This is Read 2 for the Visium v1 chemistry. |
Q30 Bases in UMI | Fraction of UMI bases with Q-score >= 30, excluding very low quality/no-call (Q <= 2) bases from the denominator. |
Fraction Reads in Spots Under Tissue | The fraction of valid-barcode, confidently-mapped-to-transcriptome reads with spot-associated barcodes. |
Total Genes Detected | The number of genes with at least one UMI count in any tissue covered spot. |
Median UMI Counts per Spot | The median number of UMI counts per tissue covered spot. |
"spaceranger count" for FFPE Metrics Definitions
The spaceranger count for FFPE pipeline outputs metrics_summary.csv which differs from spaceranger count with regard to a few key metrics.
| Metric | Description |
|---|---|
Reads Mapped to the Probe Set | Fraction of reads that map with at least one read half to the probe reference. |
Reads Mapped Confidently to the Probe Set | Fraction of reads that map uniquely with both read halves to the probe reference. |
Reads Mapped Confidently to the Filtered Probe Set | Fraction of reads that map uniquely with both read halves to the filtered probe reference. This metric will be None when probe filtering is disabled. |
Targeted Spatial Gene Expression Metrics
A Targeted Spatial Gene Expression library has additional metrics specific to targeting.
| Metric | Description |
|---|---|
Analyzed Reads | Fraction of the reads used for mapping and analysis. This fraction will be less than 100% when the sequencing depth exceeds 15,000 Mean Reads Under Tissue per Spot. See Targeted Subsampling. |
Mean Targeted Reads per Spot | The number of targeted reads, both under and outside of tissue, divided by the number of barcodes associated with a spot under tissue. |
Number of Targeted Genes | Number of targeted genes as specified in the input target panel file. |
Median Targeted Genes per Spot | The median number of targeted genes detected per tissue-associated barcode. Detection is defined as the presence of at least 1 UMI count. This metric will appear instead of the metric Median Genes per Spot above. |
Median Targeted UMI Counts per Spot | The median number of targeted UMI counts per tissue-associated barcode. This metric will appear instead of the metric Median UMI Counts per Spot above. |
Targeted Sequencing Saturation | The fraction of targeted reads originating from an already-observed targeted UMI. This is a function of library complexity and sequencing depth. More specifically, this is the fraction of confidently mapped, valid spot-barcode, valid targeted UMI reads that had a non-unique (spot-barcode, UMI, gene). This metric will appear instead of the metric Sequencing Saturation above. |
Total Targeted Genes Detected | The number of targeted genes with at least one UMI count in any spot. This metric will appear instead of the metric Total Genes Detected above. |
Number of Targeted Genes | Number of targeted genes as specified in the input target panel file. |
Number of Non-Targeted Genes | Number of genes in the reference genome that are not targeted. |
Number of Targeted Genes >= 10 UMIs | Number of targeted genes with at least 10 UMIs in spot-associated barcodes. These genes are then considered in the calculation of per-gene enrichments. |
Number of Non-Targeted Genes >= 10 UMIs | Number of non-targeted genes with at least 10 UMIs in spot-associated barcodes. These genes are then considered in the calculation of per-gene enrichments. |
Number of Enriched Targeted Genes | Number of targeted genes that are classified as enriched as a result of having high mean reads per UMI. Only genes with at least 10 spot-associated UMIs can be enriched. See Targeted Gene Expression Algorithms. |
Number of Enriched Non-Targeted Genes | Number of non-targeted genes that are classified as enriched as a result of having high mean reads per UMI. Only genes with at least 10 spot-associated UMIs can be enriched. See Targeted Gene Expression Algorithms.. |
Mean Reads per UMI per Targeted Gene | Mean number of reads per UMI per targeted gene, averaged across all targeted genes with at least 10 spot-associated UMIs. |
Mean Reads per UMI per Non-Targeted Gene | Mean number of reads per UMI per non-targeted gene, averaged across all targeted genes with at least 10 spot-associated UMIs. |
"spaceranger aggr" Metrics Definitions
The spaceranger aggr pipeline outputs summary.json that contains metrics relating to the aggregated datasets. Note: Square brackets denote a variable that depends on the pipeline input,
such as, [library_id]_frac_reads_kept means that if your aggregation contains two libraries with IDs sample123 and sample456, there are two output metrics sample123_frac_reads_kept and sample456_frac_reads_kept.
| Metric | Description |
|---|---|
filtered_bcs_transcriptome_union | The estimated number of barcodes associated with a spot under tissue, summed across all input libraries. |
total_reads | Total number of sequenced reads, summed across all input libraries. |
multi_transcriptome_total_raw_reads_per_filtered_bc | total_reads divided by filtered_bcs_transcriptome_union. |
[library_id]_pre_normalization_raw_reads_per_filtered_bc | The mean total reads per spot prior to depth normalization, for the library denoted by library_id. |
[library_id]_pre_normalization_cmb_reads_per_filtered_bc | The mean confidently mapped and barcoded (CMB) reads per spot prior to depth normalization, for the library denoted by library_id. |
[library_id]_frac_reads_kept | The fraction of reads that were retained after depth normalization for the library denoted by library_id. |
lowest_frac_reads_kept | The lowest fraction of reads retained, corresponding to the library which lost the most reads during normalization. A low value may indicate a large disparity in the initial depth of the input libraries. |
If one or more of the aggregated samples was a Targeted Gene Expression sample, these additional metrics will also appear:
| Metric | Description |
|---|---|
[library_id]_pre_normalization_targeted_reads_per_filtered_bc | The mean targeted reads per spot prior to depth normalization, for the library denoted by library_id. |
[library_id]_frac_targeted_reads_kept | The fraction of reads mapped uniquely and confidently to targeted genes that were retained after depth normalization for the library denoted by library_id. This field will be shown instead of the metric [library_id]_frac_reads_kept above. |
- 1.0
- 1.1
- 1.2
- 2.0 (latest)
- Space Ranger v1.3