10x Genomics
Chromium Single Cell Gene Expression
Cell Ranger1.3, printed on 08/08/2025
Troubleshooting Cell Ranger
When the cellranger mkfastq or cellranger count pipelines fail, they will automatically generate a "debug tarball" that contains the logs and metadata generated by the pipestance leading up to failure. This file, named sampleid.mri.tgz, can be e-mailed to the 10x software team to help resolve any issues with using Cell Ranger. You may also use the cellranger upload command to send the tarball to 10x:
$ cellranger upload your@email.edu sample_id.mri.tgz
If you wish to troubleshoot a pipeline failure yourself, it is important to identify if you are experiencing a preflight failure, an in-flight failure, or an alert.
| The remainder of this guide uses the term pipestance to refer to a specific instance of a pipeline running. |
Preflight Failures
Preflight failures are the most common and are the result of invalid input data or runtime parameters. Because they occur before the pipeline actually runs, there will be no pipeline output and the error is reported directly to your terminal.
Common preflight failures include failing to install bcl2fastq. cellranger mkfastq will generate the following error if Illumina's bcl2fastq software is not installed:
[error] On machine: workstation.university.edu, bcl2fastq or configureBclToFastq.pl not found on PATH.
In-Flight Failures
In-flight failures are generally the result of factors external to the pipeline such as running out of system memory or disk space. Different stages may fail in different ways so the specific error messages vary widely.
Finding relevant error logs
There are a few important files that are saved to your pipeline output directory which, by default, is named according to the flowcell serial number for cellranger mkfastq (e.g., HAWT7ADXX) and your --id name for cellranger count.
-
The pipeline execution log that is output to your terminal during pipeline execution is also saved to output_dir/_log.
-
Stages that experience a hard failure generate an _errors file containing the precise error that caused a stage to halt. You view these error logs, if they exist, using find output_dir -name _errors | xargs cat
-
Each stage also logs its stdout and stderr streams to _stderr and _stdout files. These logs can be listed using find output_dir -name _stderr and may contain elucidating error messages in stages that execute third-party applications such as STAR.
A more detailed description of the pipeline output directory and its contents is given in the Pipestance Structure page.
| If you are unable to diagnose a failure yourself, you can always contact the 10x software support team for help. |
Resuming a failed pipestance
Once you have determined the reason for failure and are ready to continue running the pipeline, you can typically issue the same cellranger command to continue execution of the pipestance from the stage that originally failed.
When running cellranger mkfastq or cellranger count, it will detect if its intended output directory already exists. If it does, this existing pipeline output directory will be treated as an incomplete pipestance and resume execution. This feature allows pipelines to be stopped and resumed with great flexibility, but it can also result in errors such as:
RuntimeError: /home/jdoe/runs/sample345 is not a pipestance directory
which indicates that you specified a --id that corresponds to an existing directory that was not created by Cell Ranger.
The following error:
RuntimeError: pipestance 'HAWT7ADXX' already exists and is locked by another Martian instance. If you are sure no other Martian instance is running, delete the _lock file in /home/jdoe/runs/HAWT7ADXX and start Martian again.
indicates that you may already have a copy of cellranger running that is using the same output directory. If you are sure that there is no pipestance running in the given output directory, you can either remove that output directory entirely (mv HAWT7ADXX HAWT7ADXX.old) to restart the pipestance from the beginning, or you can remove the pipestance's lock file (rm HAWT7ADXX/_lock) and re-run the cellranger command to resume pipeline execution.
If you encounter the following error when attempting to resume a pipestance:
RuntimeError: pipestance 'sample345' already exists with different invocation file /home/jdoe/runs/sample345/_invocation
you are attempting to resume a pipestance using command-line arguments that are different from those used to first run it. You can view the parameters input to the existing pipeline by examining the _log file located in the output directory (e.g., head -n20 /home/jdoe/runs/sample345/_log)
Alerts
Alerts are generally the result of factors inherent in library preparation and sequencing instead of software. Abnormal data (including common short-read sequencing metrics and Chromium-specific statistics) are raised in the form of alerts that are printed in the output web_summary.html file. Alerts do not affect the operation of the pipeline, but they do highlight potential causes for abnormal or missing data.
Alerts come in two severity levels:
-
WARN alerts indicate that some parameter is suboptimal, but there may still be useful data in the pipeline output.
-
ERROR alerts indicate a major issue, and there is unlikely to usable results in the output.
For example, running cellranger on a human sample with a human and mouse reference will likely result in an alert indicating low read alignment quality:
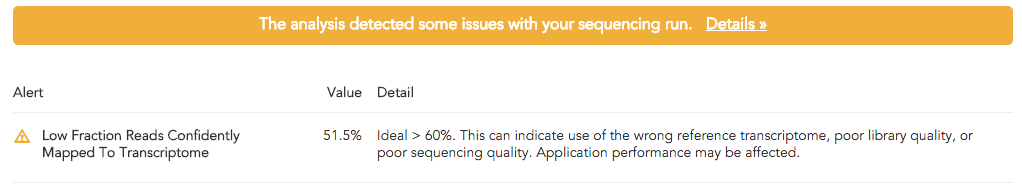
These WARN alerts are not indicative of a lost sequencing run, and re-running cellranger with the proper human reference would likely not return this alert.
Poorly constructed libraries typically display more severe ERROR alerts. For example, a low-quality library may result in the following alerts:
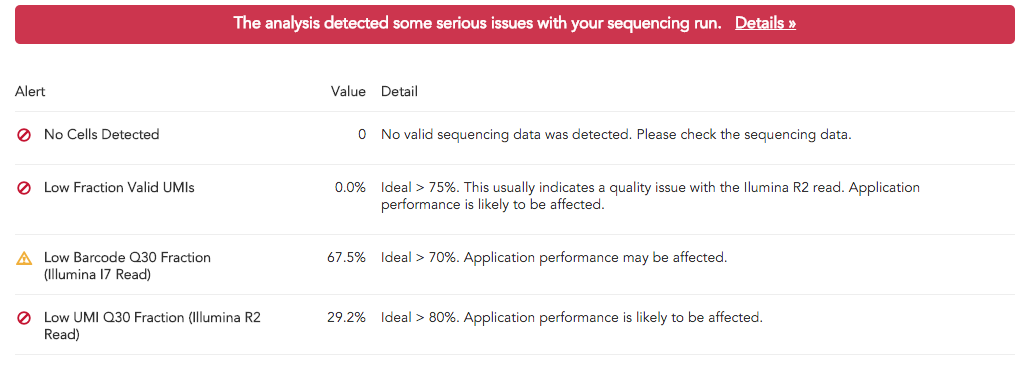
The presence of these ERROR alerts indicate that results that are output by this pipestance are likely dubious.