10x Genomics
Chromium Single Cell Immune Profiling
Cell Ranger7.1 (latest), printed on 07/09/2025
Running Cell Ranger aggr
Table of Contents
- What is aggr?
- Aggregating outputs from Cell Ranger vdj
- Aggregating outputs from Cell Ranger multi
- Aggregating outputs from a combination of Cell Ranger vdj and Cell Ranger multi
- Aggregating outputs from Cell Ranger count
- GEM well suffix in the barcode
What is aggr?
Many experiments involve generating data for samples that are processed through different Gel Bead-in-emulsion (GEM) wells on the Chromium instrument. Depending on the experimental design, these could be replicates from the same set of cells, cells from different tissues or time points from the same individual, or cells from different individuals. The cellranger count, cellranger vdj, and cellranger multi pipelines process data from a single GEM well. The aggr pipeline aggregates the outputs from multiple runs of cellranger count/vdj/multi and performs analysis on the combined data.
| cellranger aggr is not designed to aggregate multiple sequencing runs of the same library from the same GEM well (e.g., resequencing samples to increase sequence depth). For this scenario, instead of running cellranger aggr, run a single analysis of either cellranger count, vdj, or multi and specify the list of FASTQ files from each sequencing run of the same GEM well with the fastqs field. |
Aggregating outputs from Cell Ranger vdj
The cellranger aggr command takes a CSV file specifying a list of cellranger vdj output files (specifically the vdj_contig_info.pb from each run) and performs clonotype grouping on the aggregated data. Consider two instances of B cell vdj pipeline using the sequencing data from two separate GEM wells prepared using the 10x Genomics Chromium platform, as described in this section.
cd /opt/runs cellranger vdj --id=Lib1 ... ... wait for pipeline to finish ... cellranger vdj --id=Lib2 ... ... wait for pipeline to finish ...
To aggregate these datasets, you need to create a CSV containing the following columns:
| Column Name | Description |
|---|---|
sample_id | Unique identifier for this input GEM well. This will be used for labeling purposes only; it does not need to match any previous ID assigned to the GEM well. |
vdj_contig_info | Path to the contig info file produced by cellranger vdj. For example, if you processed your GEM well by calling cellranger vdj --id=ID in some directory /DIR, this path would be /DIR/ID/outs/vdj_contig_info.pb |
donor | See the Glossary |
origin | See the Glossary |
How are donor and origin values used in aggr?
There are three ways Cell Ranger can process the datasets depending on the combination of donor and origin values:
- If two datasets come from the same donor but have different origins, Cell Ranger will rerun the clonotype grouping algorithm on the combined set of cells. This allows cells from different datasets to belong to the same clonotype.
- If two datasets come from the same donor and origin, then Cell Ranger performs additional filtering to remove certain rare artifacts. For example, Cell Ranger will filter expanded exact subclonotypes that are present in one library but not in another from the same origin, which would be highly improbable, assuming random draws of cells from the tube. These are believed to arise when a plasma or plasmablast cell breaks up during or after pipetting from the tube, and the resulting fragments contaminate GEMs, yielding expanded false clonotypes that are residues of real single plasma cells.
- If two cells came from different donors, then Cell Ranger will not put them in the same clonotype.
In addition to these CSV columns, cellranger aggr accepts additional columns containing library meta-data (e.g., vaccination status). These custom library annotations do not affect the analysis pipeline but can be visualized downstream in the Loupe V(D)J Browser.
You can either make the CSV file in a text editor or create it in Excel and export it as a CSV file. Continuing the example from the previous section, your Excel spreadsheet would look like this:
| A | B | C | D | E | |
|---|---|---|---|---|---|
| 1 | sample_id | vdj_contig_info | donor | origin | VaccinationStatus |
| 2 | Sample1 | /opt/runs/Sample1/outs/vdj_contig_info.pb | D1 | pbmc_t0 | Pre-Vaccination |
| 3 | Sample2 | /opt/runs/Sample2/outs/vdj_contig_info.pb | D1 | pbmc_t1 | Post-Vaccination |
Required columns and experimental design information are shown here:
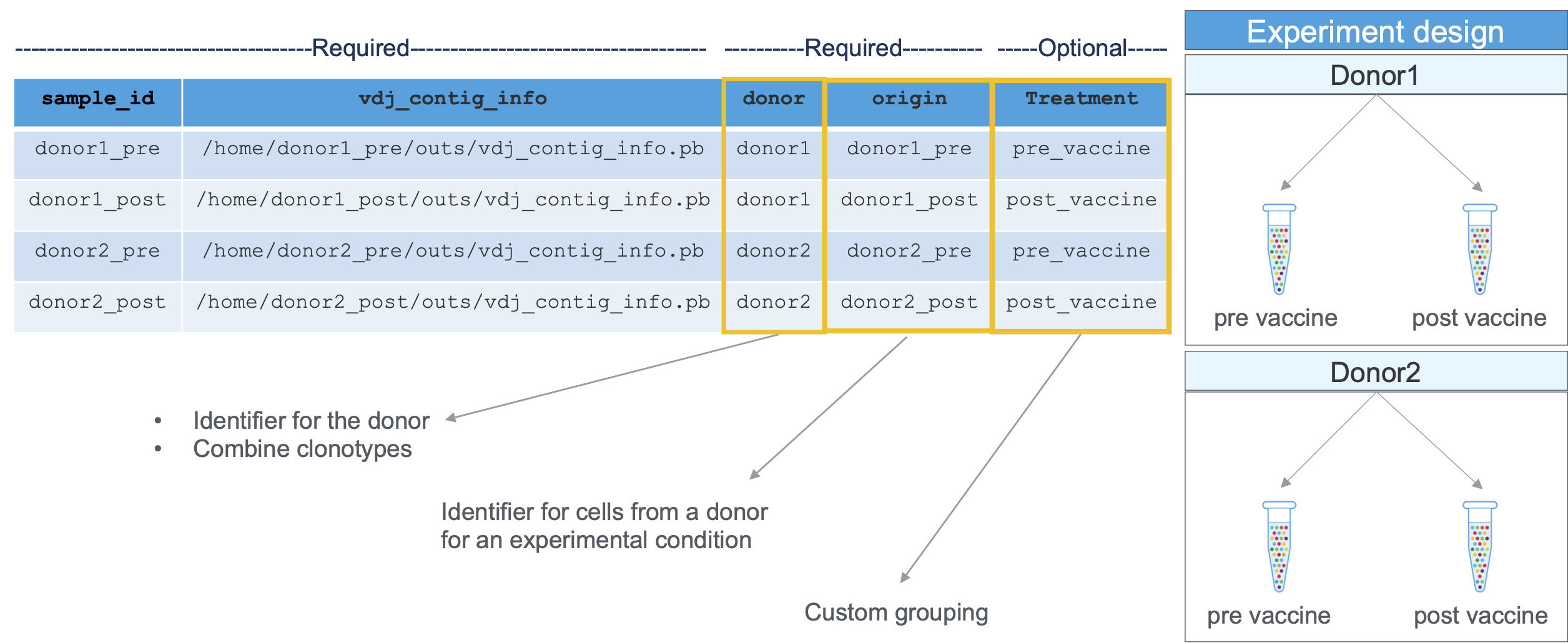
When you save it as a CSV, the result would look like this:
sample_id,vdj_contig_info,donor,origin,VaccinationStatus Sample1,/opt/runs/Lib1/outs/vdj_contig_info.pb,D1,pbmc_t0,Pre-Vaccination Sample2,/opt/runs/Lib2/outs/vdj_contig_info.pb,D1,pbmc_t1,Post-Vaccination
You can run the aggr pipeline as follows:
cd /opt/runs cellranger aggr --id=pre_post_vac_aggr --csv=aggr.csv
The pipeline will produce an error if the individual libraries were run using different V(D)J references or if the chain type (TCR or IG) in the individual libraries is inconsistent.
A successful run should conclude with a message similar to this:
- Copy of the input aggregation CSV: /opt/runs/pre_post_vac_aggr/outs/aggregation.csv
- count: null
- vdj_t: null
- vdj_b:
Aggregation metrics summary HTML: /opt/runs/pre_post_vac_aggr/outs/vdj_b/web_summary.html
Clonotypes csv: /opt/runs/pre_post_vac_aggr/outs/vdj_b/clonotypes.csv
Clonotype consensus FASTA: /opt/runs/pre_post_vac_aggr/outs/vdj_b/consensus.fasta
Annotations of filtered contigs with library metadata (CSV): /opt/runs/pre_post_vac_aggr/outs/vdj_b/filtered_contig_annotations.csv
Clonotype consensus annotations (CSV): /opt/runs/pre_post_vac_aggr/outs/vdj_b/consensus_annotations.csv
- V(D)J reference:
fasta:
regions: /opt/runs/pre_post_vac_aggr/outs/vdj_reference/fasta/regions.fa
donor_regions: /opt/runs/pre_post_vac_aggr/outs/vdj_reference/fasta/donor_regions.fa
reference: /opt/runs/pre_post_vac_aggr/outs/vdj_reference/reference.json
Pipestance completed successfully!
Each output file produced by cellranger aggr follows the format described in the Understanding Outputs section of the documentation, and it includes the union of all the relevant barcodes from each input jobs. The GEM well suffix of each barcode is updated to prevent barcode collisions, as described above.
Once cellranger aggr has completed, browse the resulting summary HTML file in any supported web browser, open the .vloupe file in Loupe V(D)J Browser, or refer to the other output files to explore the data:
- clonotypes.csv
- consensus.fasta
- filtered_contig_annotations.csv: We also include the donor, origin and any custom metadata specified in the input aggregation CSV as additional columns in this csv file.
- consensus_annotations.csv
- donor_regions.fa
Aggregating outputs from Cell Ranger multi
The cellranger aggr command can take a CSV file specifying a list of cellranger multi output directories, and perform aggregation on any combination of 5' Gene Expression, Antibody Capture, CRISPR, and V(D)J libraries that are present in the individual runs of cellranger multi.
Cell Ranger aggr cannot be used to aggregate Antigen Capture libraries. If an Antigen Capture library is present in your analysis, cellranger aggr runs into a preflight error:
[error] Aggr with Antigen Capture libraries is unsupported.
However, all the runs of cellranger multi included in the aggregation CSV must have the same combination of libraries. For example, if Sample1 has Gene Expression, BCR, and TCR, whereas Sample2 has only Gene Expression and BCR, cellranger aggr fails with this error message:
[error] Pipestance failed. Error log at: path/to/error_message/chnk0-u4933b35480/_errors Log message: The multi outs folders supplied as inputs to aggr contain an inconsistent set of libraries: - 'Sample1' contains [count, vdj_t, vdj_b] - 'Sample2' contains [count, vdj_b] To aggregate multi outputs, all the individual inputs must contain the same set of library types. If you would like to use aggr for only one library type, you can supply the respective `molecule_info` (for count) or the `vdj_contig_info` (for vdj)
Consider two multi datasets containing data from 5' Gene Expression and V(D)J libraries:
cd /opt/runs cellranger multi --id=Sample1 --csv=exp1.csv ... wait for pipeline to finish ... cellranger multi --id=Sample2 --csv=exp2.csv ... wait for pipeline to finish ...
To aggregate these datasets, you need to create a CSV containing these columns:
| Column Name | Description |
|---|---|
sample_id | Unique identifier for this input GEM well. This will be used for labeling purposes only; it does not need to match any previous ID assigned to the GEM well. |
sample_outs | Path to the `per_sample_outs` folder generated by cellranger multi. For example, if you processed your GEM well by calling cellranger multi --id=ID --csv=exp1.csv in some directory /DIR , and the sample was called Sample1, this path would be /DIR/ID/outs/per_sample_outs/Sample1 |
donor | See the Glossary. This section describes how donor would affect the clonotype grouping in aggr. |
origin | See the Glossary. This section describes how origin would affect the clonotype grouping in aggr. |
Note that the column header vdj_contig_info is replaced by sample_outs for cellranger multi runs.
The cellranger aggr pipeline will auto-detect the presence of various libraries based on the structure and contents of the per sample outs folders. Apart from the change in the input CSV column (sample_outs instead of molecule_h5 or vdj_contig_info), the sections on aggregating outputs from cellranger vdj and aggregating outputs from cellranger count (depth normalization, batch correction, etc.) apply here as well.
In addition to the CSV columns described above, cellranger aggr accepts optional columns that may contain additional meta-data (e.g., vaccination status). These custom library annotations do not affect the analysis pipeline but can be visualized downstream in the Loupe V(D)J Browser.
You can either make the CSV file in a text editor or create it in Excel and export it as a CSV. Your Excel spreadsheet might look like this:
| A | B | C | D | E | |
|---|---|---|---|---|---|
| 1 | sample_id | sample_outs | donor | origin | VaccinationStatus |
| 2 | Sample1 | /opt/runs/Sample1/outs/per_sample_outs/Sample1 | D1 | pbmc_t0 | Pre-Vaccination |
| 3 | Sample2 | /opt/runs/Sample2/outs/per_sample_outs/Sample2 | D1 | pbmc_t1 | Post-Vaccination |
When you save it as a CSV, the result would look like this:
sample_id,sample_outs,donor,origin,VaccinationStatus Sample1,/opt/runs/Sample1/outs/per_sample_outs/Sample1,D1,pbmc_t0,Pre-Vaccination Sample2,/opt/runs/Sample2/outs/per_sample_outs/Sample2,D1,pbmc_t1,Post-Vaccination
You can run the aggr pipeline as follows:
cd /opt/runs cellranger aggr --id=pre_post_vac_aggr --csv=aggr.csv
Aggregating outputs from a combination of Cell Ranger vdj and Cell Ranger multi
The cellranger aggr command can be used to aggregate outputs from a combination of cellranger multi and cellranger vdj runs. The aggregation CSV must have the column names described in the Aggregating outputs from Cell Ranger vdj section.
The path to the vdj_contig_info.pb file is required even for samples processed with cellranger multi. The vdj_contig_info.pb is located in the sample_outs/ directory with a path similar to /per_sample_outs/Sample1/vdj_b/vdj_contig_info.pb for a BCR library.
This example CSV is used to aggregate the outputs from a BCR library processed with cellranger multi and a BCR library processed with cellranger vdj
sample_id,vdj_contig_info,donor,origin Sample1,/opt/runs/Sample1/outs/per_sample_outs/Sample1/vdj_b/vdj_contig_info.pb,D1,pbmc_t0 Sample2,/opt/runs/Sample2/outs/vdj_contig_info.pb,D1,pbmc_t0,D1,pbmc_t0
Aggregating outputs from Cell Ranger count
The following page will take you to relevant sections on aggregating outputs from cellranger count (gene expression and Feature Barcode data). While the page is hosted on the 3' Single Cell solution's section, they are equally applicable to the 5' Immune Profiling solution as well:
GEM well suffix in the barcode
Each GEM well is a physically distinct set of GEM partitions that draws barcode sequences randomly from the pool of valid barcodes cataloged in the barcode whitelist. To track barcodes when aggregating multiple libraries, Cell Ranger appends an integer identifying the GEM Well to the barcode nucleotide sequence and uses that nucleotide sequence and ID as the unique identifier. For example, AGACCATTGAGACTTA-1 and AGACCATTGAGACTTA-2 are distinct cell barcodes from different GEM wells, despite having the same barcode nucleotide sequence.
This number, which specifies which GEM well the barcode sequence originated from, is called the GEM well suffix. The numbering of the GEM wells reflects the order that the GEM wells were provided in the aggregation CSV.