10x Genomics
Chromium Single Cell Immune Profiling
Cell Ranger6.0, printed on 07/28/2025
Analyzing V(D)J and Gene Expression / Feature Barcode with cellranger multi
The 5' Chromium Single Cell Immune Profiling Solution with Feature Barcode technology enables simultaneous profiling of V(D)J repertoire, cell surface protein, antigen specificity and gene expression data. The cellranger multi pipeline enables the analysis of these multiple library types together. The advantage of using the multi pipeline (as opposed to using cellranger vdj and cellranger count separately) is that it enables more consistent cell calling between the V(D)J and gene expression data. This involves the following steps:
-
Run cellranger mkfastq on the Illumina BCL output folder to generate FASTQ files.
-
Run cellranger multi on FASTQ files produced by cellranger mkfastq.
Run cellranger mkfastq
First, follow the instructions on running cellranger mkfastq to generate FASTQ files. For example, if the flowcell serial number was HAWT7ADXX, then cellranger mkfastq will output FASTQ files in HAWT7ADXX/outs/fastq_path.
Run cellranger multi
To simultaneously generate single-cell feature counts, V(D)J sequences and annotations for a single library, run cellranger multi. This requires a config CSV, which is described below, and invoking cellranger multi with the following arguments:
| Argument | Description |
|---|---|
--id | A unique run ID string: e.g. sample345 that is also the output folder name. Cannot be more than 64 characters. |
--csv | Path to config CSV file enumerating input libraries and analysis parameters. |
The multi config CSV contains both the library definitions and experiment configuration variables. It is composed of up to four sections: [gene-expression], [feature], [vdj], and [libraries]. The [gene-expression], [feature], and [vdj] sections have at most two columns, and are responsible for configuring their respective portions of the experiment. The [libraries] section specifies where input FASTQ files may be found. A template for a multi config CSV can be downloaded here, and example multi config CSVs can be downloaded from 5.0.0 public datasets here.
| Multi Config CSV | |
|---|---|
Section: [gene-expression] | |
| Field | Description |
reference | Path of folder containing 10x-compatible reference. Required for gene expression and Feature Barcode libraries. |
target-panel | Optional. Path to a target panel CSV file or name of a 10x Genomics fixed gene panel (pathway, pan-cancer, immunology, neuroscience). |
no-target-umi-filter | Optional. Disable targeted UMI filtering stage. Default: false. |
r1-length | Optional. Hard trim the input Read 1 of gene expression libraries to this length before analysis. Default: do not trim Read 1. |
r2-length | Optional. Hard trim the input Read 2 of gene expression libraries to this length before analysis. Default: do not trim Read 2. |
chemistry | Optional. Assay configuration. NOTE: by default, the assay configuration is detected automatically, which is the recommended mode. Users usually will not need to specify a chemistry. Options are: 'auto' for autodetection, 'threeprime' for Single Cell 3', 'fiveprime' for Single Cell 5', 'SC3Pv1' or 'SC3Pv2' or 'SC3Pv3' for Single Cell 3' v1/v2/v3, 'SC5P-PE' or 'SC5P-R2' for Single Cell 5', paired-end/R2-only, 'SC-FB' for Single Cell Antibody-only 3' v2 or 5'. Default: auto. |
expect-cells | Optional. Expected number of recovered cells. Default: 3000. |
force-cells | Optional. Force pipeline to use this number of cells, bypassing cell-calling algorithm. |
include-introns | Optional. Include intronic reads in count. Default: false |
no-secondary | Optional. Disable secondary analysis, e.g. clustering. Default: false. |
no-bam | Optional. Do not generate a bam file. |
Section: [feature] | |
| Field | Description |
reference | Optional. Feature reference CSV file, declaring Feature Barcode constructs and associated barcodes. Required only if Feature Barcode libraries are present. |
r1-length | Optional. Hard trim the input Read 1 of Feature Barcode libraries to this length before analysis. Default: do not trim Read 1. |
r2-length | Optional. Hard trim the input Read 2 of Feature Barcode libraries to this length before analysis. Default: do not trim Read 2. |
Section: [vdj] | |
| Field | Description |
reference | Path of folder containing 10x-compatible VDJ reference. Required for VDJ Immune Profiling libraries. |
inner-enrichment-primers | Optional. If inner enrichment primers other than those provided in the 10x kits are used, they need to be specified here as a text file with one primer per line. |
r1-length | Optional. Hard trim the input Read 1 of V(D)J libraries to this length before analysis. Default: do not trim Read 1. |
r2-length | Optional. Hard trim the input Read 2 of V(D)J libraries to this length before analysis. Default: do not trim Read 2. |
Section: [libraries] | |
| Column | Description |
fastq_id | The Illumina sample name to analyze. This will be as specified in the sample sheet supplied to mkfastq or bcl2fastq. Required. |
fastqs | The folder containing the FASTQ files to be analyzed. Generally, this will be the fastq_path folder generated by cellranger mkfastq. Required. |
lanes | Optional. The lanes associated with this sample, separated by `|`. Defaults to using all lanes. |
feature_types | The underlying feature type of the library, which must be one of ‘Gene Expression’, ‘VDJ’, ‘VDJ-T’, ‘VDJ-B’, ‘Antibody Capture’, or ‘CRISPR Guide Capture’. Required. |
subsample_rate | Optional. The rate at which reads from the provided FASTQ files are sampled. Must be strictly greater than 0 and less than or equal to 1. |
For help on how to configure the [libraries] section to target a particular set of FASTQs, consult Specifying Input FASTQ Files for cellranger multi.
|
After determining these input arguments, run cellranger:
$ cd /home/jdoe/runs $ cellranger multi --id=sample345 --csv=/home/jdoe/sample345.csv
Following a series of checks to validate input arguments, cellranger multi pipeline stages will begin to run:
Martian Runtime - v4.0.4 Running preflight checks (please wait)... ...
By default, cellranger will use all of the cores available on your
system to execute pipeline stages. You can specify a different number of cores
to use with the --localcores option; for example, --localcores=16
will limit cellranger to using up to sixteen cores at once. Similarly,
--localmem will restrict the amount of memory (in GB) used by
cellranger.
The pipeline will create a new folder named with the run ID you specified using the --id argument (e.g. /home/jdoe/runs/sample345) for its output. If this folder already exists, cellranger will assume it is an existing pipestance and attempt to resume running it.
Output Files
A successful cellranger multi run should conclude with a message similar to this:
Waiting 6 seconds for UI to do final refresh. Pipestance completed successfully! yyyy-mm-dd hh:mm:ss Shutting down. Saving pipestance info to "tiny/tiny.mri.tgz"
The output of the pipeline will be contained in a folder named with the run ID you specified (e.g. sample345). The subfolder named outs will contain the main pipeline outputs:
Upon completion, the cellranger multi pipeline will produce an outs directory with the following structure:
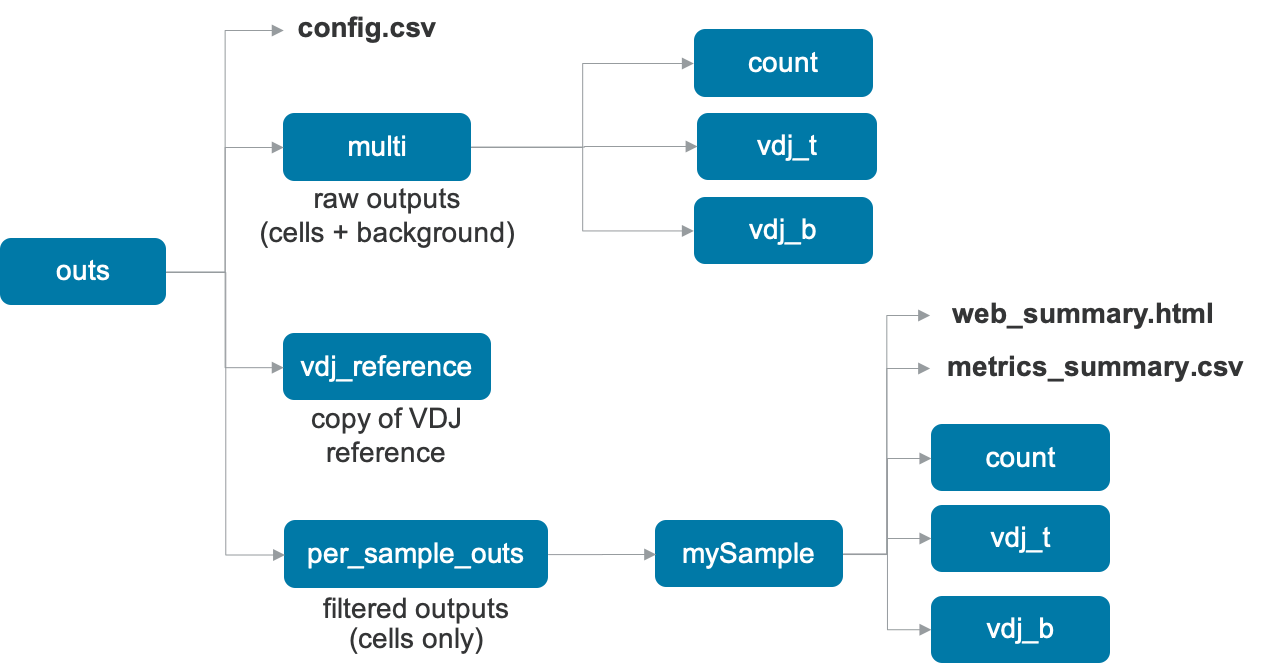
The files in the multi folder contain raw data, i.e., the data for all barcodes including cells and background, while the files in the per_sample_outs directory are the filtered files, i.e. for cells in this sample. The pipeline also outputs a copy of the input VDJ reference in the vdj_reference directory.
| File Name | Description |
|---|---|
web_summary.html | Run summary metrics and charts in HTML format |
metrics_summary.csv | Run summary metrics in CSV format |
config.csv | The input multi config CSV |
count | The results of any gene-expression and feature barcode analysis, similar to cellranger count, described here |
vdj_b | The results of any V(D)J Immune Profiling analysis for any B cells, similar to cellranger vdj, described here |
vdj_t | The results of any V(D)J Immune Profiling analysis for any T cells, similar to cellranger vdj, described here |
Once cellranger multi has successfully completed, you can browse the resulting summary HTML file in any supported web browser, or refer to the count and vdj sections to explore the data by hand.
When to use the multi pipeline
| VDJ | 5' GEX | 5' FB | Use multi? |
|---|---|---|---|
| Yes | Yes | Yes | Recommended |
| Yes | Yes | No | Recommended |
| Yes | No | Yes | Optional. No effect on cell calling |
| Yes | No | No | Optional |
| No | No | Yes | Optional |
| No | Yes | No | Optional |
| No | Yes | Yes | Optional |
The gene expression library is representative of the entire pool of poly-adenylated mRNA transcripts captured within each partition (droplet). The TCR or BCR transcripts are then selectively amplified to create the V(D)J library. Therefore, the gene expression library has more power to detect partitions containing cells compared to the V(D)J library. If the multi pipeline is run with both gene expression and VDJ data, then barcodes which are not called as cells by using the gene expression data will be deleted from the V(D)J cell set.
Features not in cellranger multi
The --denovo option available in cellranger vdj is not supported in cellranger multi.
Additional features in cellranger multi
The cellranger multi pipeline supports downsampling the reads by specifying a rate between 0 and 1 independently for each library. It also allows trimming the reads to a fixed length, which is not supported in the cellranger vdj pipeline.