10x Genomics
Chromium Single Cell CNV
Cell Ranger DNA1.0, printed on 08/02/2025
Pipestance Structure
The pipeline output directory, described in Understanding Output, contains all of the data produced by one invocation of a pipeline (a pipestance) as well as rich metadata describing the characteristics of each stage. This directory contains a specific structure that is used by the Martian pipeline framework to track the state of the pipeline as execution proceeds.
Pipeline Structure
Cell Ranger DNA's notion of a pipeline is very flexible in that a pipeline can be composed of stages that run stage code or sub-pipelines that may themselves contain stages or sub-pipelines.
Cell Ranger DNA pipelines follow the convention that stages are named with verbs (e.g., TRIM_READS, MARK_DUPLICATES, DETECT_BREAKPOINTS) and sub-pipelines are named with nouns and prefixed with an underscore (e.g., _BCSORTER). Each stage runs in its own directory bearing its name, and each stage's directory is contained within its parent pipeline's directory.
For example, the cellranger-dna mkfastq pipeline has the following process graph:
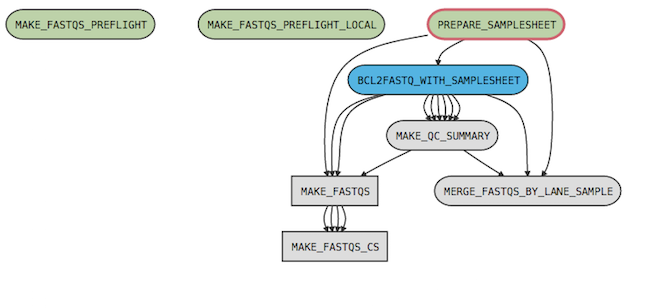
where
- MAKE_FASTQS_CS is the top-level pipeline stage
- MAKE_FASTQS is a sub-pipeline contained in MAKE_FASTQS_CS
- PREPARE_SAMPLESHEET, BCL2FASTQ_WITH_SAMPLESHEET, MAKE_QC_SUMMARY, and MERGE_FASTQS_BY_LANE_SAMPLE are stages contained in the MAKE_FASTQS sub-pipeline.
- MAKE_FASTQS_PREFLIGHT and MAKE_FASTQS_PREFLIGHT_LOCAL are preflight stages, which validate inputs prior to running the other stages. These also belong to MAKE_FASTQS, but have no connections to other stages because they don't produce any outputs.
| The MAKE_FASTQS_CS stage is not strictly necessary since it contains no stages and only one child pipeline (MAKE_FASTQS); however, it serves to mask some of the low-level inputs required by the MAKE_FASTQS pipeline. |
Directory Structure
Every pipestance operates wholly inside of its pipeline output directory. When the pipestance completes, this pipestance output directory contains three outputs: metadata files, the pipestance output file directory, and the top-level pipeline stage directory.
- Metadata files are files prefixed with an underscore (_) and usually contain unstructured text or JSON-encoded arrays and hashes.
- The pipestance output file directory is a directory called outs/ that contains the pipestance's output files.
- The top-level pipeline stage directory is a directory named according to the top-level pipeline stage that contains the child stage directories that compose this pipestance.
The top-level pipeline stage directory is a stage directory that contains any number of child stage directories as well as one stage output directory for each fork run by that stage. There are four possible top-level pipeline stages:
- MAKE_FASTQS_CS for cellranger-dna mkfastq
- CNV_CALLER_SINGLECELL_CS for cellranger-dna cnv
All of the Cell Ranger DNA pipelines only contain single-fork stages, so there will only ever be a fork0 stage output directory within each stage directory. Chunk output directories are a subset of stage output directories that additionally contain runtime information specific to the job or process being run by that chunk (e.g., a process ID or cluster job ID).
For example, the Cell Ranger DNA mkfastq pipeline's pipeline output directory contains the following directory structure:
| _log | Metadata file |
| outs/ | Pipestance output file directory |
| MAKE_FASTQS_CS/ | Top-level pipeline stage directory |
| MAKE_FASTQS_CS/fork0/ | Stage output directory |
| MAKE_FASTQS_CS/fork0/files/ | Stage output files |
| MAKE_FASTQS_CS/MAKE_FASTQS/ | Stage directory |
| MAKE_FASTQS_CS/MAKE_FASTQS/fork0/ | Stage output directory |
| MAKE_FASTQS_CS/MAKE_FASTQS/fork0/files/ | Stage output files |
| MAKE_FASTQS_CS/MAKE_FASTQS/BCL2FASTQ_WITH_SAMPLESHEET/ | Stage directory |
| MAKE_FASTQS_CS/MAKE_FASTQS/BCL2FASTQ_WITH_SAMPLESHEET/fork0/ | Stage output directory |
| MAKE_FASTQS_CS/MAKE_FASTQS/BCL2FASTQ_WITH_SAMPLESHEET/fork0/chnk0/ | Chunk output directory |
Commonly Generated Metadata
The metadata contained in the pipeline output directory includes
| File Name | Description |
|---|---|
| _finalstate | Metadata cache that is populated when a pipestance completes to minimize re-aggregation of metadata |
| _invocation | The MRO call used to invoke this pipestance |
| _log | The log messages that are reported to your terminal window when running cellranger-dna commands |
| _mrosource | The entire MRO describing the pipeline with all @include statements dereferenced |
| _perf | Detailed runtime performance data for every stage in the pipestance |
| _timestamp | The start and finish time for this pipestance |
| _vdrkill | A list of all of the volatile data (temporary files) removed during pipeline execution as well as total number of files and bytes deleted |
| _versions | Versions of the components used by the pipeline |
Stage directories contain stage output directories, stage output files, and the stage directories of any child stages or pipelines.
Stage output directories typically contain:
| File Name | Contents |
|---|---|
| files/ | Directory containing any files created by this stage that were not considered volatile (temporary) |
| split/ | A special stage output directory for the step that divided this stage's input into parallel chunks |
| chnkN/ | A chunk output directory for the Nth parallel chunk executed |
| join/ | A special stage output directory for the step that recombined this stage's parallel output chunks into a single output dataset again |
| _complete | A file that, when present, signifies that this stage has successfully completed |
| _errors | A file that, when present, signifies that this stage failed. Contains the errors that resulted in stage failure. |
| _invocation | The MRO call used to execute this stage by the Martian framework |
| _outs | The output files generated by this stage |
| _vdrkill | A list of all of the volatile data (temporary files) removed during pipeline execution as well as total number of files and bytes deleted |
Chunk output directories are a subset of stage output directories that, in addition to the aforementioned stage output, may contain:
| File Name | Contents |
|---|---|
| _args | The arguments passed to the stage's stage code |
| _jobinfo | Metadata describing the stage's execution, including performance metrics, job manager jobid and jobname, and process ID |
| _jobscript | The script submitted to the cluster job manager (cluster mode-only) |
| _stdout | Any stage code output that was printed to the stdout stream |
| _stderr | Any stage code output that was printed to the stderr stream |
These metadata files should be treated as read-only, and altering the contents of metadata files is not recommended.
Navigating Pipestances
Pipestance output directories can demonstrate very complicated structures, and re-attaching the Cell Ranger DNA UI is the easiest way to quickly navigate to a pipeline stage of interest and examine its metadata. In the absence of being able to access the UI, the standard find command can quickly return high-level information about a pipestance.
For example, to find the stages that resulted in the overall failure of a pipestance whose output directory is sample345/,
$ find sample345/ -name _errors sample345/CNV_CALLER_SINGLECELL_CS/CNV_CALLER_SINGLECELL/_BREAKPOINT_PIPELINE/AGGREGATE_NODES/fork0/join-u82692d3c96/_errors
This tells us that the failed stage was AGGREGATE_NODES.
It can be helpful to view all _errors files' contents at once by piping to xargs cat:
$ find sample345/ -name _errors | xargs cat Traceback (most recent call last): File "/home/jdoe/cellranger-dna-1.0.0/martian-cs/v3.0.0/adapters/python/martian_shell.py", line 541, in _main stage.main() File "/home/jdoe/cellranger-dna-1.0.0/martian-cs/v3.0.0/adapters/python/martian_shell.py", line 513, in main self._run(lambda: self._module.join( File "/home/jdoe/cellranger-dna-1.0.0/martian-cs/v3.0.0/adapters/python/martian_shell.py", line 476, in _run cmd() File "/home/jdoe/cellranger-dna-1.0.0/martian-cs/v3.0.0/adapters/python/martian_shell.py", line 514, inargs, outs, chunk_defs, chunk_outs)) File "/home/jdoe/cellranger-dna-1.0.0/cellranger-dna-cs/1.0.0/mro/stages/cluster_breakpoints/aggregate_nodes/__init__.py", line 225, in join assert X.shape[1] == Y.shape[1] AssertionError
In the above case, the error is an unhandled exception whose cause is not obvious; these sorts of failures should be reported to the 10x software support team for assistance with diagnosis.
Stages whose stage code run external binaries (for example, the ALIGN stage which runs BWA) often generate output to their stdout and stderr streams. These messages are captured in the _stdout and _stderr metadata files within the chunk output directories, and combining find and xargs cat to examine their contents can also assist with troubleshooting.
- 1.1 (latest)
- Cell Ranger DNA v1.0